Switzerland relies significantly on imported medical devices. With the implementation of the EU Medical Devices Regulation (MDR) and a mutual recognition agreement (MRA) terminated in May 2021, the availability of medical devices compliant with the EU laws on the Swiss territory is decreasing. To avoid a sudden shortage of medical devices, in November 2022, the Swiss Parliament requested the recognition of medical devices compliant with other highly regulated systems. The Parliament instructed the Swiss Federal Council to adapt the national laws to accept FDA-approved medical devices.
As the Swiss legislation aligns with the EU law, in August 2023, Swiss Medtech published an Expert Report from the Johner Institut that analyses the differences and similarities between the US and EU regulatory systems for medical devices. The report takes into account several aspects such as conformity assessments, safety and performance requirements, and market surveillance, comparing FDA-approved products with CE-marked medical devices.
Centralised vs. decentralised regulatory systems
In the United States, the Federal Food, Drug, and Cosmetic Act (FD&C Act) regulates medical devices. The FDA, the U.S. Food and Drug Administration, monitors the implementation and application of the regulatory requirements. The FDA is also responsible for the approval and market surveillance of medical devices. Consequently, the USA has a very centralised regulatory system, that relies on internal experts and high transparency.
In the European Union, the European Parliament and the European Council enact the regulatory framework for medical devices. In all 27 Member States and EEA countries, the Medical Devices Regulation (MDR) applies since May 2021. Unlike the US, the EU does not have a centralised regulatory system. As opposed to the American system, the EU single countries are responsible for market surveillance in their territory, while notified bodies, private third-party bodies, are in charge of the conformity assessment of high-risk products.
Key differences: scope and classification
When comparing the two laws, one main difference arises when looking at the definition of “medical device”. The American legislation defines as medical devices products also for veterinary use, whereas the MDR definition includes only products for human use and leaves veterinary products out of scope. Regarding the classification, both the US and EU laws have a classification system based on risk classes:
- In the United States, medical devices are classified from class I to class III, where class I includes low-risk devices, class II is for moderate-risk devices, and class III devices are products that play an essential role in the patient’s vital functions. Manufacturers can research the class of their devices with a product code to enter into an FDA-dedicated databank.
- In the European Union, the classification of medical devices considers, among others, the product type, the application type, as well as the duration and place of application. Going from low risk to the highest risk, medical devices are classified from class I to class III, where class I also differentiates between measure products (class Im), sterile products (class Is), and reusable surgical products (class Ir). Manufacturers are responsible for the classification. However, notified bodies and competent authorities can verify and challenge the classification given.
If comparing, the United States has the advantage that the FDA can easily adjust the classification of certain devices when market data prove a different risk assessment. As a result, the US can react more easily to new technologies.
FDA approval procedure and EU conformity assessment of medical devices
To enter the market, medical devices in both the United States and the European Union have to undergo a compliance process.
In the United States, there are three types of approval processes:
- Premarket Notification, or 510(k), which applies to product obtaining approval based on substantial equivalences with other existing devices (known as “predicate devices”). This process typically covers devices posing moderate risk (class II) or rarely class I products.
- De Novo Classification Request, where the manufacturer has to prove the safety and performance of the devices through clinical data, performance evaluation and other information, including biocompatibility, sterility, and cyber security. This process is chosen when there are no predicate devices to compare the product with.
- Premarket Approval (PMA) is the approval process for class III medical devices for which compliance with the “general and special controls” set by the law is not sufficiently proven to ensure the safety and performance of the device. The manufacturer has to provide scientific evidence, in particular clinical evidence, to prove that the product is safe and effective. On average, the FDA approves 31 medical devices through this procedure annually.
Most class I and some class II devices that the FDA classifies as “uncritical” do not need administrative approval and none of the above-described procedures applies. In this category, there are also many software products. The FDA lists such products with exempt status in a dedicated databank.
In the EU, medical devices must be CE-marked to be placed on the market. Class I devices that are not class Im, Ir, and Is, can bear the CE marking when the manufacturer declared conformity with the applicable requirements (Declaration of Conformity or “DoC”). For all other classes, including class Im, Ir, and Is, manufacturers can affix the CE marking only following a conformity assessment procedure performed by a notified body. To summarise, an overview of the EU conformity assessment procedures:
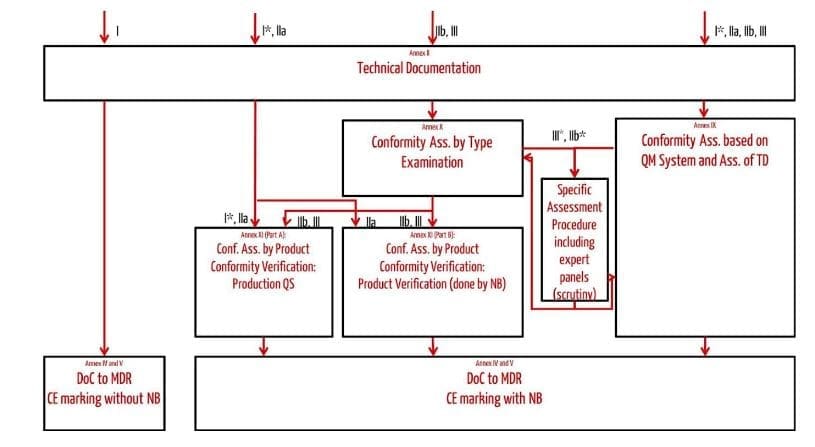
Source: Johner Institut (2023)
US and EU systems equally safe
In conclusion, the Expert Report of the Johner Institut does not underline any differences between the two systems in relation to safety aspects. Medical devices are highly regulated in both the US and EU. There are overall many common aspects in both systems and differences relate only to specificities.
Despite lacking a Mutual Recognition Agreement with the EU, Switzerland still recognises CE-marked devices and will soon add FDA-approved medical devices in their national supply.
Check our Library of Documents dedicated to regulatory compliance in Switzerland.
References:
Johner Institut (2023) Vergleich der regulatorischen Systeme hinsichtlich der Sicherheit von Medizinprodukten aus den USA und der EU. Retrieved on 05.09.2023.
Leave a Reply