The MDCG issued a Questions & Answers (Q&A) document aimed at assisting sponsors by clarifying certain aspects of clinical investigation of medical devices within the bounds of the Regulation (EU) 2017/745 (MDR).
This document provides a more detailed overview concerning general subjects such as definitions of relevant concepts, regulatory pathways, responsibilities, and other specific requirements of clinical investigations. The Q&A also refers to modifications to clinical investigations, timelines, reports, and arrangements for the transitional period. In the end, a clear overview of the regulatory pathway for clinical investigation under MDR can be found as well as a non-exhaustive list of modifications that may be interpreted as substantial.
Below some of the Questions & Answers from the new MDCG document:
1. What are the general differences and improvements related to clinical investigations under the new Regulation (EU) 2017/745 (MDR) as compared to the Directives 93/42/EEC and 90/385/EEC?
Regulation (EU) 2017/745 (MDR) will progressively replace both Directives (93/42/EEC and 90/385/EEC) and their transpositions in national law.
The first difference is regarding the type of the law. A Directive is a legislative act that sets out a goal that all EU countries must achieve. However, it is up to the individual countries how to reach these goals by the implementation of national laws. A Regulation, as opposed to a Directive, is a binding legislative act that must be applied in its entirety across the EU on the date of application. It means that the rules are applied in an identical manner throughout the EU. Member States, in authorising and supervising the conduct of a clinical investigation, will be required to base their assessments and decisions on the same rules.
The MDR contains greater detail than the Directive, which is a result of implementing aspects related to good clinical practice, many of which have previously been present in the form of guidance and standard documents.
Further harmonisation at European level will provide greater certainty, which will support an environment that provides greater predictability and is more favourable for conducting clinical investigations, with the highest standards of patient safety, for all EU Member States. It will not only harmonise decisions, but also foster work sharing and collaboration between Member States and enhance the transparency regarding these studies.
For certain clinical investigations (Article 82(2) of the MDR, Article 70(7)(a) of the MDR) the sponsor still needs to check and follow any specific national provisions which may apply.
10. Who is responsible for determining the correct regulatory pathway for a clinical investigation?
It is the sponsor’s responsibility to determine the correct regulatory pathway for their clinical investigation. Guidance is provided in this document, but the MDR and national legislation contain the legally binding requirements.
11. What are the safety reporting requirements for clinical investigations?
The requirements for safety reporting will depend on whether you are using the investigated medical device within its intended purpose:
• If the investigated medical device is CE marked and will be used within its intended purpose, the provisions on vigilance laid down in Article 80(5) and Articles 87 to 90 of the MDR and the acts adopted pursuant to Article 91 of the MDR shall apply for PMCF clinical investigations.
• If the investigated medical device is not CE marked, or is CE marked but will be used outside its intended purpose, the provisions on safety reporting laid down in Article 80of the MDR shall apply.
Please refer to MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for further guidance’
26. The clinical investigation module in Eudamed will not be ready by May 2021, how can the sponsor follow the regulation without this functionality in Eudamed?
Clinical investigation sponsors will not have the opportunity to be recorded in Eudamed as of the date of application of the MDR.
To submit an application:
All requested information to apply for or notify a clinical investigation should be submitted to the national competent authorities unless otherwise specified in the MS concerned. Check with the relevant National Competent Authority which system will be used for submission. The Commission has a listing of the contact details for National Competent Authorities, which is available at: https://ec.europa.eu/health/sites/health/files/md_sector/docs/md_clinical_investigation_contact_points.pdf
To fulfil the safety reporting requirements of MDR Article 80:
Please see MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for guidance.
27. What will happen to those clinical investigations that started prior to the date of application of Regulation (EU) 2017/745?
Clinical investigation that are currently being conducted with respect to Directive 93/42/EC and Directive 90/385/EC by the date of application of the MDR, can continue to be conducted. Nevertheless, serious adverse events (SAEs) and device deficiencies occurring after the date of application of the MDR, shall be notified to the MS according to the rules defined in Article 80 of the MDR.
To facilitate the transition and give time for sponsors to update Clinical Investigation Plans and procedures in clinical investigations a sponsor may continue to report all SAEs to National Competent Authorities until Eudamed reporting is mandatory. This applies only to studies which have started to be conducted in accordance with Article 10 of Directive 90/385/EEC or Article 15 of Directive 93/42/EEC prior to 26 May 2021. Please refer to MDCG 2020-10/1 ‘Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745’ for further guidance.
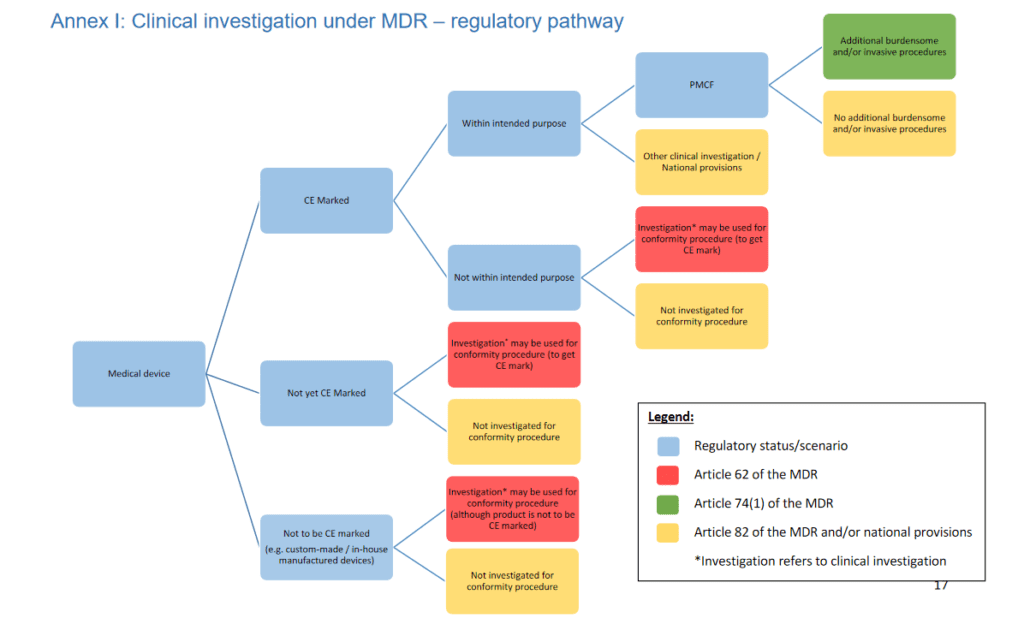
Annex I: Clinical investigation under MDR – regulatory pathway
You can find all Questions & Answers here as well as in our Library of documents by clicking on MDCG or Clinical Evaluation!
Leave a Reply